Description
Required Software
Gaussian09
AmberTools 12 or above
Overview
Usage
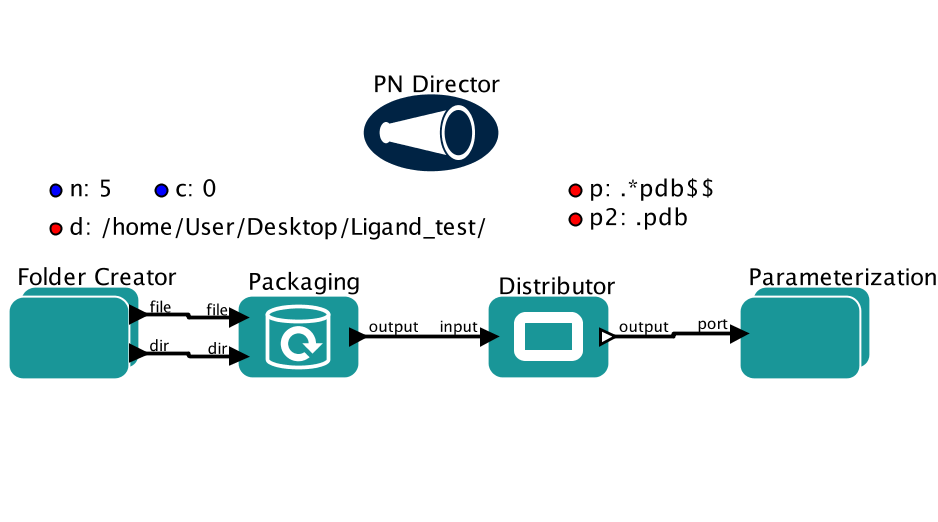
Parameters
c charge of the small molecules. All the small molecules must have the same charges. Default is 0 whichmeans all the small molecules are neutral.
d directory with the small molecule pdb files.
p pattern of the pdb files. Default is .*pdb$$ which means all the pdb files in the directory.
p2 pattern of the pdb file to be replaced by empty string in order to obtain only the ligand name. Defaultis .pdb which means that LIGAND_NAME.pdb will turn into LIGAND_NAME
n the number of pdbs to parameterize in a single run. For example, if user has five small moleculesin a single directory that need to parameterize, n will be set to five.