Description
Required Software
AmberTools 12 or 13
Overview
Usage
Parameters
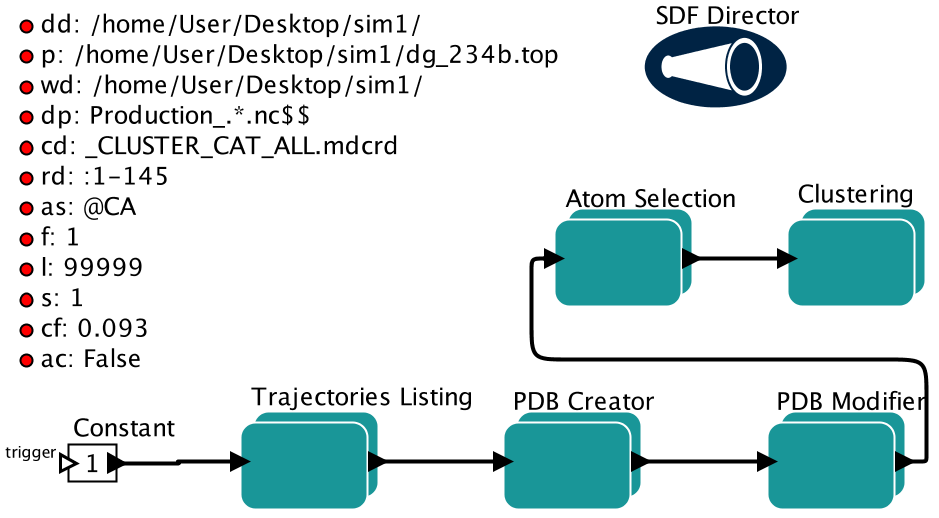
dd Trajectories directory. This should be the full path to the trajectories directory.
dp Trajectories pattern. Pattern examples: .*pdb$$ (all pdb files in the trajectories directory),.*nc$$ (all nc files), .*mdcrd$$ (all mdcrd files), .*dcd$$ (all dcd files), .+size.*$$ (all files thatcontain size in the middle), and Size.*$$ (all files that contains Size in the begining)
p Location of the topology file. This should be a full path to the topology file.
wd Path of the work directory. This is where all the intermediate files and output files are stored.
cd Name of the trajectory file which will contain all of the input trajectories. Default name is_CLUSTER_CAT_ALL.mdcrd
rd Residue id. When creating the input for gromos clustering, user can choose to keep only certain trajectorystructures. For example, if a user wants to keep the protein and strip everything else in a trajectory,the user will need to specify the protein residue id that are intended to keep. This naming conventionfollows Amber mask convention. For example, if the protein in the trajectories is residue numbers 1 to 194,users will input :1-194.
as Atom selection. This flag specifies which atoms or residues that clustering will be based on. Default valueis all alpha carbons. This flag naming convention follows Amber mask convention.
f First frame. Users can choose to have a set number of frames for gromos clustering. Default value is thefirst frame.
l Last frame. Users can choose to have a set number of frames for gromos clustering. Default value is 999999999.
s Stride. Users can choose to have a set number of frames for gromos clustering. Default value is 1, no stride.2 will mean every other frame.
cf Cutoff. This is the RMSD cutoff for gromos clustering. A very small rmsd cutoff will cause the program to fail.
ac True or False. If true, alignment will be done on alpha carbon. If false, alignment will be done on the atomsselected also for clustering. Default is False.